Role of Machine Learning in Molecular Discovery & Scientific Understanding
The exponential growth of computational power combined with the emergence of innovative machine learning (ML) algorithms offers a revolutionary paradigm in the realm of molecular discovery and scientific understanding. This workshop aims to create a dialogue between pure ML researchers, and scientists using cutting-edge models for data-centric scientific molecular and material discovery.
Happening soon!
 Joint workshop on the Role of Machine Learning in Molecular Discovery & Scientific Understanding
Joint workshop on the Role of Machine Learning in Molecular Discovery & Scientific Understanding
Key note speaker
Prof. dr. Max Welling
Research Chair Machine Learning, University of Amsterdam
Date: 21 March 2024
Venue: Theatre Hall X, TU Delft (directions)
Full programme (Pdf) - now available
Schedule:
09.30 - walk in with coffee
10.00 - morning session
12.00 - lunch
13.00 - afternoon sessions
17.00 - drinks & bites
Registration form - registration has closed (for questions, please contact r.boerrigter@tudelft.nl)
Participation is free. If you registered we expect you to be present.
We will share some practical information with everyone that registered prior to the workshop.
Scope
The workshop encompasses the principles and advancements in machine learning and their applications in material and molecular discovery. It also emphasizes the role of machine learning in the prediction of crystal structures, the discovery of stable functional molecules, and the rational material design process. The challenges in integrating ML/AI with first-principle simulations, the formation of new collaborative networks and partnerships, and the importance of interpretability and knowledge extraction in ML models will also be discussed.
Join us
Whether your interest lies in developing machine learning algorithms or using them to unlock the mysteries of the molecular world, we believe this workshop will offer invaluable insights. Join us in this exploration of the future of scientific discovery, facilitated by the powerful combination of machine learning and quantum mechanics.
Programme
It promises to be a dynamic intersection of pure machine learning and statistical models applied to scientific discovery. The event will focus on exploring theoretical machine learning research and their real-world applications in molecular and material discovery.
The emphasis will be on integrating small scale experimental data and quantum mechanical calculations into machine learning frameworks, forming collaborative networks, and addressing key challenges.
Prof. dr. Max Welling - keynote speaker

Title: Relating Non-Equilibrium Thermodynamics to Machine Learning: what can we learn?
Full Professor and Research Chair Machine Learning
University of Amsterdam / AMLAB
Max Welling is a fellow at the Canadian Institute for Advanced Research (CIFAR) and the European Lab for Learning and Intelligent Systems (ELLIS) where he serves on the founding board.
His previous appointments include Partner and VP at Microsoft Research, VP at Qualcomm Technologies, professor at UC Irvine, postdoc at UCL & U. Toronto under supervision of prof. Geoffrey Hinton, and postdoc at Caltech under supervision of prof. Pietro Perona. He finished his PhD in theoretical high energy physics under supervision of Nobel laureate prof. Gerard ‘t Hooft.
Max Welling has served as associate editor in chief of IEEE TPAMI from 2011-2015, he serves on the advisory board of the Neurips foundation since 2015, he is co-founder of the European Lab for Learning and Intelligence Systems (ELLIS) and served on its board until 2021, he has been program chair and general chair of Neurips in 2013 and 2014 respectively. He was also program chair of AISTATS in 2009 and ECCV in 2016 and general chair and co-founder of MIDL 2018.
He is recipient of the ECCV Koenderink Prize in 2010 and the ICML Test of Time award in 2021.
@wellingmax (X)
Confirmed speakers
- Prof. dr. Max Welling, University of Amsterdam (Key note speaker)
Title: Relating Non-Equilibrium Thermodynamics to Machine Learning: what can we learn?
* * Unfortunately, Dr. Süleyman Er will not be able to present today. We updated the programme. * *
Dr.  Süleyman Er
Süleyman Er
Head of the Solar Fuels Department, DIFFER
Group Leader of Autonomous Energy Materials Discovery, DIFFER
Title: Towards Autonomous Discovery of Energy Materials
Abstract
In this talk, I will present an overview of our research projects that merge AI with computational screening for identifying electrolyte molecules, which can potentially be used for energy storage in aqueous redox flow batteries. Our approach is multi-faceted, involving: I) Development of theoretical methodologies [1,2] and AI tools [3] to evaluate redox potentials and solubilities of candidate compounds in water [4,5], along with automated chemical space visualization [6] and chemical price search tools for sourcing these molecules from suppliers. II) Investigation of structure-property relationships and prioritization of compounds, aiming to pinpoint the most effective candidates for practical experiments [7,8]. III) Conducting electrochemical tests on safe and easily accessible compounds selected from our virtual library [7]. IV) The creation of a computational database of electroactive molecules [9]. These examples demonstrate how AI and computational science can be coupled to provide guidance in the search for potential energy materials.
References
[1] Q. Zhang, A. Khetan, S. Er, Scientific Reports 10, 22149 (2020). https://doi.org/10.1038/s41598-020-79153-w
[2] Q. Zhang, A. Khetan, S. Er, Scientific Reports 11, 4089 (2021). https://doi.org/10.1038/s41598-021-83605-2
[3] M.C. Sorkun, E.N. Ghassemi, C. Yatbaz, J.M.V.A. Koelman, S.Er, ChemRxiv (2023). https://doi.org/10.26434/chemrxiv-2023-2ncbf
[4] M.C. Sorkun, A. Khetan, S. Er, Scientific Data 6, 143 (2019). https://doi.org/10.1038/s41597-019-0151-1
[5] M.C. Sorkun, J.M.V.A. Koelman, S. Er, iScience 24, 101961 (2021). https://doi.org/10.1016/j.isci.2020.101961
[6] M.C. Sorkun, D. Mullaj, J.M.V.A. Koelman, S. Er, Chemistry-Methods 2, e202200005 (2022). https://doi.org/10.1002/cmtd.202200005
[7] Q. Zhang, A. Khetan, E. Sorkun, F. Niu, A. Loss, I. Pucher, S. Er, Energy Storage Materials 47, 167 (2022). https://doi.org/10.1016/j.ensm.2022.02.013
[8] Q. Zhang, A. Khetan, E. Sorkun, S. Er, Journal of Materials Chemistry A 10, 22214 (2022). https://doi.org/10.1039/D2TA05674G
[9] E. Sorkun, Q. Zhang, A. Khetan, M.C. Sorkun, S. Er, Scientific Data 9, 718 (2022). https://doi.org/10.1038/s41597-022-01832-2
Short bio | Süleyman Er
Süleyman Er is leading both the Solar Fuels Department and the Autonomous Energy Materials Discovery Group at DIFFER. His academic background in Chemistry and Physics has paved the way for his research focused on the development and application of computational and AI methods to accelerate the discovery of molecules and materials for a variety of energy conversion and storage applications. His achievements include numerous research grants, projects, publications, patents, AI-based software tools, and FAIR databases of molecules and materials. He has been honored with several research and innovation awards, such as the Young Energy Scientist and eScience Center Fellowships, along with recognition in the Mission Innovation Champions Program, and the William A. Goddard Medal. Er has also supervised a diverse cadre of researchers, many of whom have successfully advanced in their careers across various sectors.
Dr.  Jana Weber
Jana Weber
Assistant professor, Delft University of Technology, Delft Bioinformatics Lab, Faculty of Electrical Engineering, Mathematics and Computer Science
Title: Towards generative design of molecular ensembles with machine learning
Abstract
Synthetic polymers are a highly demanded class of materials that one finds in many different consumer products. AI-assisted in-silico design of molecules is becoming an increasingly valuable approach to accelerate molecular discovery and development, yet generative AI for synthetic polymers still needs to overcome domain-specific challenges. One challenge is that unlike for small molecules, synthetic polymers are governed by multiple structural levels of information, beyond the atomistic structure of monomers. This causes challenges for data collection and for machine-readable representations alike. Secondly, targeted, or controlled design of new materials requires much (property-) labelled data, which in the field of synthetic polymers is not yet easily accessible. In this talk, I will present our current works on molecular machine learning for copolymers. We build upon the representation of polymers as molecular graph ensembles [1] and work on the two challenges outlined above: learning with limited labelled data and learning beyond the atomistic representation of monomer units [2].
References
[1] Aldeghi, M., & Coley, C. W. (2022). A graph representation of molecular ensembles for polymer property prediction. Chemical Science, 13(35), 10486-10498.
[2] Vogel, G., Sortino, P., & Weber, J. M. (2023). Graph-to-String Variational Autoencoder for Synthetic Polymer Design. In AI for Accelerated Materials Design-NeurIPS 2023 Workshop.
Short bio | Jana Weber
Jana M. Weber is an assistant professor for artificial intelligence in bioscience at TU Delft in the Department of Intelligent Systems since 2022. She manages the AI4b.io lab and she is part of the Delft Bioinformatics Lab. In 2022, Jana defended her PhD in Chemical Engineering from the University of Cambridge for her work on circular chemistry through large scale reaction network analysis and optimization. Prior to that, Jana obtained her MSc in Environmental (Process) Engineering from RWTH Aachen University with her Master’s thesis performed at the Jülich Research centre at the institute of bio- and geosciences. Jana’s research interests cover molecular machine learning and network science for a broad range of environmental applications. In particular, she focuses on molecular property prediction, inverse design, and on utilising biochemical reaction data for identifying more sustainable reaction pathways.
 Pim de Haan
Pim de Haan
PhD Student, University of Amsterdam
Senior engineer Qualcomm AI Research
Title: Scientific machine learning with the Geometric Algebra Transformer
Abstract
Problems involving geometric data arise in physics, chemistry, robotics, computer vision, and many other fields. The geometric (or Clifford) algebra is a vector space that can encode such geometric data, and it can elegantly express powerful geometric computations. In this talk, I will introduce the Geometric Algebra Transformer (GATr), a general-purpose architecture that uses the geometric algebra as its inputs, outputs, and hidden states. The architecture is equivariant to the symmetries of space and therefore sample-efficient. Also, it is scalable to large problems, as it uses the same optimized computational kernels as regular transformers. I will discuss the benefits of GATr in various scientific applications, including the estimation of wall-shear-stress in human arteries.
Short bio | Pim de Haan
Pim de Haan studied theoretical physics at the universities of Amsterdam and Cambridge, and now pursues a PhD in machine learning at the University of Amsterdam, supervised by Max Welling. Also, he is a researcher at Qualcomm AI Research. His research interests are in applications of machine learning to geometric, interactive and scientific problems, utilizing the structure and symmetries of the domains.
 Dr. Menno Bokdam
Dr. Menno Bokdam
Assistant Professor, University of Twente, Faculty of Science & Technology and MESA+ Institute for Nanotechnology
Title: Looking inside ‘dynamic solids’ using on-the-fly machine learning force fields
Abstract
Ab intio based Molecular Dynamics (MD) is known for its high accuracy, but also for its unfeasible demand for computational power when going beyond one thousand atoms and picosecond time scales. In recent years, several important approaches have been developed that have enabled a very successful application of machine learning in this field. These advances are so substantial that it could mean "the end of ab initio MD"*. I will introduce our implementation of on-the-fly learning in a commonly used Density Functional Theory software package, and focus on the potential that the method has for research in condensed matter physics. Examples from the (in)organic halide perovskites where we have simulated observables that would have been impossible using conventional methods will be presented. The common denominator of a much larger class of ‘dynamic solids’ is their anharmonic lattice dynamics and wide variety of rare-events. Large thermodynamic ensembles must be simulated with MD to obtain, for example: phase transition temperatures and melting points, phonon bandstructures, X-ray spectra and thermal conductivity. We observe a qualitative and quantitative agreement between experiment and theory on several temperature depended NMR experimental observables. This leads us to suspect that the simulated trajectories of crystal structure provide a realistic insight into the ionic dynamics of these perovskites.
* G. Csányi, Quote from keynote, Psi-K conference ‘22
Short bio | Menno Bokdam
Menno Bokdam is an ab-initio simulations expert in solid state physics & chemistry, focusing on finite temperature atomic structure prediction of ‘Dynamic Solids’ and structure-property relations. He is a co-developer of the on-the-fly Machine-Learning Force Fields (MLFF) method, integrated in the ab-initio package VASP. Using MLFF molecular dynamics he simulated the thermodynamic structural phase space, including phase transitions and related transition pathways, of the prime examples of 3D organic perovskites. After his PhD at the University of Twente (UT), Bokdam gained six years of research experience at the Physics faculty of the University of Vienna. In 2020 he re-joined the UT as a tenure track assistant professor, with a research line focusing on the development of MLFF measurements of experimental observables, and applications relevant for the semiconductor industry. He teaches courses in Machine Learning, Hilbert Spaces and Electronic Structure Theory and develops FAIR compliant research data visualization software.
 Dr. Jakub Tomczak
Dr. Jakub Tomczak
Associate Professor and PI of the Generative AI Group, Eindhoven University of Technology
Title: Generative AI with Decision Making for Drug Design
Abstract
ChatGPT and Stable Diffusion showed that Generative AI is the way to go in building a new generation of AI systems. But what about other applications, beyond images, text and audio? What about the tractability of deep generative models? In this talk, we will focus on two aspects: molecular generation and property prediction. Furthermore, we will discuss whether current models are tractable and if not, whether we can formulate tractable deep generative models for drug design.
Short bio | Jakub Tomczak
Jakub M. Tomczak is an associate professor and the PI of the Generative AI group at the Eindhoven University of Technology (TU/e). Before joining the TU/e, he was an assistant professor at Vrije Universiteit Amsterdam, a deep learning researcher (Engineer, Staff) in Qualcomm AI Research in Amsterdam, a Marie Sklodowska-Curie individual fellow in Prof. Max Welling's group at the University of Amsterdam, and an assistant professor and a postdoc at the Wroclaw University of Technology. His main research interests include deep generative modeling, deep learning, and Bayesian inference, with applications to image/text processing, Life Sciences, and Molecular Sciences. He serves as an action editor of "Transactions of Machine Learning Research", and an area chair of major AI conferences (e.g., NeurIPS, ICML, AISTATS). He is the author of the book entitled "Deep Generative Modeling", the first comprehensive book on Generative AI. He is also the founder of Amsterdam AI Solutions.
 Dr. Will Robinson
Dr. Will Robinson
Postdoc, The Huck Group, Institute for Molecules and Materials, Radboud University, and Senior Group Leader in the RobotLab
Title: Machine Learning’s role in understanding chemical reaction systems
Abstract
Living systems are admired for their abilities to grow, adapt and evolve in complex environments. Their grasp of how to harness the mixed chemical and energetic resources available from their surroundings is key to performing these feats, and a huge inspiration for chemists. The chemistry of Life can be thought of as a set of processes which control chemical compositions using interconnected systems of reactions. Understanding how to mimic these systems in the absence of sophisticated cellular machinery is a huge challenge. To perform and understand chemistry on the systems level will enable new classes of biomimetic reactions which can not only provide reflective insight into their biological counterparts, but also access to a range of chemical technologies which may provide efficient means to harness challenging chemical resources such as biomass and waste streams.
Investigating chemical reaction systems requires the collection of large amounts of multivariate data which must be processed and analysed for compositional and behavioural trends. In this talk, I will present an overview of the data analysis and machine learning methods we have employed in understanding reaction pathway self-organisation in the formose reaction, a model prebiotic reaction network (Nat. Chem., 2022, 14, 623-631), as well as how this reaction may be used for classification and time-series prediction tasks as a reservoir computer. Our experiment-led work has adopted machine learning methods as a matter of course. I am looking forward to showing you how we are using them to enable our ambitious projects, as well as a forward-looking view into how we are going to combine machine learning and chemical data in the new RobotLab.
Short bio | Will Robinson
As a PhD candiate Will Robinson studied enzyme electrochemistry and the mechanism of CO2 reduction by formate dehydrogenase in the group of Prof. Erwin Reisner (University of Cambridge, Department of Chemistry). As postdoc he focussed on developing semi-artificial photosynthetic systems which efficiently couple photo-driven water oxidation to CO2 reduction to formate and proton reduction to formate.
For the last few years, Will Robinson been working in the group of Prof. Wilhelm Huck (Radboud University Nijmegen) on understanding how systems of chemical reactions self-organise. This work began in the realm of prebiotic chemistry, in which we developed insight into how chemical reactivity and environmental conditions combine to create order in chemical reaction systems without sophisticated cellular machinery. It has now evolved into using chemical reactions to process information and perform classification and predictive tasks. Applying machine learning methods has become essential in enabling this challenging work.
 Dr. Nong Artrith
Dr. Nong Artrith
Assistant Professor, Materials Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University
Title: Machine Learning (ML) for Simulating Complex Energy Materials with Non-Crystalline Structures
Abstract
Many materials with applications in energy, e.g., batteries, are non-crystalline, exhibiting amorphous structures, chemical disorder, and complex compositions. This complexity makes direct modeling with first principles methods challenging. To address this challenge, we developed accelerated sampling strategies based on machine learning interatomic potentials, genetic algorithms, and molecular-dynamics simulations [1]. Here, I will discuss the methodology and its applications to amorphous battery materials. We constructed the phase diagram of amorphous LiSi alloys, which are prospective anode materials for lithium-ion batteries [2]. Additionally, we mapped the composition and structure space of amorphous lithium thiophosphate (LPS) solid electrolytes [3-5]. The thermodynamic stability and ionic conductivity of the non-crystalline phases were correlated with local structural motifs, leading to the identification of structure-composition-conductivity relationships that can be used for materials optimization and design. X-ray absorption spectroscopy (XAS) characterizes materials, revealing details of the absorber atom’s local chemistry. Our work created an S/P K-edge XAS spectra database for LPS materials using structures from [3]. This study presents the initial atomic-scale insights into the oxidative degradation of LPS electrolytes, guiding macroscopic reactions via microstructural engineering and enhancing sulfide electrolyte design.
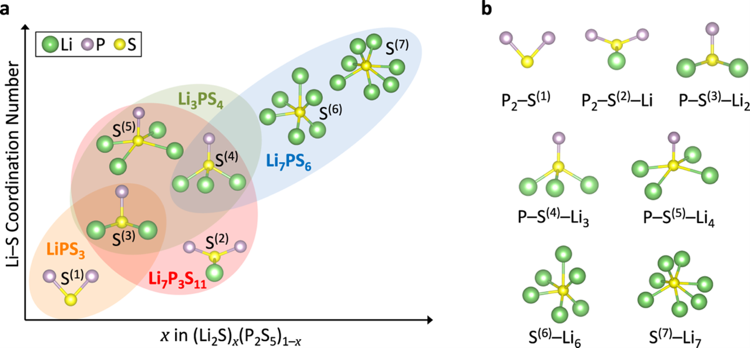
Figure 1: Schematic illustration [4] of the local coordination of S atoms with Li and P atoms in selected (Li2S)x(P2S5)1–x crystalline structures. Li: green; S: yellow; P: purple. LiPS3: orange region; Li7P3S11: red region; Li3PS4: green region; Li7PS6: blue region.
References
[1] N. Artrith and A. Urban, Comput. Mater. Sci. 114, 135-150 (2016).
[2] N. Artrith, A. Urban, and G. Ceder, J. Chem. Phys. 148, 241711 (2018).
[3] H. Guo, Q. Wang, A. Urban, and N. Artrith, Chemistry of Materials 34, 6702-6712 (2022).
[4] H. Guo, M.R. Carbone, C. Cao, N. Artrith, A. Urban, D. Lu, et al, Scientific Data 10, 349 (2023).
[5] C. Cao, M.R. Carbone, C. Komurcuoglu, S. Jagriti, S. Yoo, N. Artrith, A. Urban, D. Lu, F. Wang et al, https://doi.org/10.48550/arXiv.2310.00794
Short bio | Nong Artrith
Nong Artrith is an Assistant Professor in the Materials Chemistry and Catalysis Group at the Debye Institute for Nanomaterials Science, Utrecht University, and a Visiting Researcher at Microsoft Research Amsterdam Lab (2022-2023). Prior to joining Utrecht University, Nong was a Research Scientist at Columbia University, USA, and a PI in the Columbia Center for Computational Electrochemistry. Nong obtained her PhD in Theoretical Chemistry from Ruhr University Bochum, Germany, for the development of machine-learning (ML) models for materials chemistry. She was awarded a Schlumberger Foundation fellowship for postdoctoral research at MIT and subsequently joined UC Berkeley as an associate specialist. In 2019, Nong has been named a Scialog Fellow for Advanced Energy Storage. Since 2023, Nong is a member of the NL ARC CBBC. She is the main developer of the open-source ML package ænet for atomistic simulations. Her research interests focus on the development and application of first principles and ML methods for the computational discovery of energy materials and for the interpretation of experimental observations.
 Dr. Sid Kumar
Dr. Sid Kumar
Assistant Professor Mechanics Materials Computing, Department of Materials Science & Engineering, Faculty of Mechanical Engineering, Delft University of Technology
Title: Inverse design of materials with generative modeling: from molecules to meta-molecules
Abstract
Machine learning (ML) and specifically, generative modeling is rapidly accelerating how we inverse design materials for targeted and exotic properties (e.g., stress-strain response). However, in application-centric contexts, a purely generative approach wherein the entire material design or representation is generated may not be required or useful. Prior physical knowledge about the material system can enable breaking down the design space into coupled/uncoupled sub-spaces and in turn, accelerate the learning and discovery process. E.g., a polymer chemistry based on acid and epoxide molecules may motivate both independent and simultaneous optimization over their respective design spaces. This becomes particularly challenging if the individual design representations are extremely dissimilar (e.g., topological parameters vs. geometrical parameters), multi-modal (e.g., text- vs. graph-based), high-dimensional, and discrete and discontinuous (e.g., ad-hoc lattices and molecules). To address these challenges, we introduce a generative ML framework that constructs a low-dimensional, continuous latent representation unifying an enormous range of material designs; the latent space contains both independent and shared dimensions for both coupled and uncoupled generative modeling of individual constituents. Leveraging this framework, we inverse design
- self-healing vitrimer polymer chemistries consisting of acid and epoxide molecules for tailored glass transition temperature [1],
- ultra-light lattice-based metamaterials (with topologically and geometrically engineered unit cells or “meta-molecules” of lengthscales much larger than a molecule, yet much smaller than the bulk material) for tailored stress-strain response [2].
References
[1] Zheng et al., Inverse design of vitrimeric polymers by molecular dynamics and generative modeling, Arxiv: 2312.03690.
[2] Zheng et al., Unifying the design space and optimizing linear and nonlinear truss metamaterials by generative modeling, Nature Communications, 14, 7563 (2023).
Short bio | Sid Kumar
Sid Kumar is an Assistant Professor at TU Delft in the Department of Material Science and Engineering of the Faculty of Mechanical Engineering since 2021, where he leads the Mechanics, Materials, and Computing group. He obtained his PhD in Aeronautics from Caltech followed by a postdoc position at ETH Zürich. Previously, he obtained a dual MSc from Caltech and Ecole Polytechnique (France). Sid has been awarded the Dutch Research Council (NWO) Veni award, the Foster and Coco Stanback fellowship in Engineering and Applied Science at Caltech, and the University of Paris Saclay fellowship at Ecole Polytechnique. His research interests lie at the intersection of mechanics of materials, computational modeling, and machine learning — with a focus on inverse problems in material design and modeling.
 Prof. dr. Vedran Dunjko, Professor in Quantum Computing, Leiden University
Prof. dr. Vedran Dunjko, Professor in Quantum Computing, Leiden University
Title: Towards quantum machine learning for quantum physical systems with provable advantages
Abstract
Quantum machine learning (QML) has mostly been investigated for its potential to outperform classical machine learning methods in typical "big-data" applications. However, arguably, the natural application of is in fact in problems where genuinely quantum features matter, such as quantum mechanical systems. In this talk we will reflect on recent results proving QML can indeed provide provable advantages in quantum tasks, e.g. quantum chemistry and condensed-matter physics.
Vedran Dunjko | Short bio
Vedran Dunjko’s research interest lies in the intersection of computer science and quantum physics, including quantum computing and quantum cryptography. Over the course of the last few years, he has been focusing on the interplay between quantum computing, machine learning, and artificial intelligence. He is professor of quantum computing at Leiden University, and lead of the applied Quantum algorithms Initiative.
Personal website Vedran Dunjko
Quantum@LIACS
 Dr. Kevin Rossi will shortly introduce DAEMON COST, the European network on Machine Learning for materials design.
Dr. Kevin Rossi will shortly introduce DAEMON COST, the European network on Machine Learning for materials design.
Kevin Rossi is Assistant Professor in AI for materials design at the Materials Science and Engineering Department, Faculty of Mechanical Engineering, TU Delft, and Materials Flagship leader at the Climate Safety and Security Centre at the TU Delft | The Hague campus.
@_RossiKevin (X)
Header image: Kumar et. al., 2020 | Frame: AI generated image